

The Food And Drug Administration (FDA) approved the Pfizer-BioNTech vaccine for children as young as 5 years old despite the fact that its Pfizer-connected advisory committee knew about numerous adverse effects that were found in Pfizer’s clinical trials for children, including adverse effects that were determined to be “related” to the clinical vaccine trial. The Briefing Packet for the FDA advisory committee meeting shows that the FDA advisers used clinical studies sponsored by BioNTech and conducted by or supported by Pfizer to approve the vaccine for young children. Talk about a conflict of interest! NATIONAL FILE reported on the FDA advisory committee’s massive Pfizer connections and how committee members have worked for Pfizer and are making money from the Pfizer vaccine. So what kind of adverse effects were discovered in the clinical trials for children? The advisory committee meeting briefing packet, published by the FDA, shows that clinical trials found “related” illnesses including Lymphadenopathy, arthralgia, paresthesia, nervous tic, hematochezia (characterized by bloody stool, which sent the participant to the “emergency department”) pyrexia (fever), neutropenia (low white blood cell count), hypersensitivity reaction, angioedema, and rashes. Meanwhile, a case of the blood vessel disorder Henoch-Schoenlein purpura was reported but was conveniently classified as “non related” to the vaccine experiment.
Here is the “Vaccines and Related Biological Products Advisory Committee Meeting October 26, 2021 Briefing Document” published by the FDA. Page 15 lists an “OVERVIEW OF CLINICAL STUDIES.” Two clinical studies are listed. The first clinical study listed is “Phase 1/2/3 Registrational Study C4591001,” which was sponsored by BioNTech and conducted by Pfizer. The other clinical study listed is “Phase 1/2/3 Pediatric Study C4591007,” which is listed as being sponsored by BioNTech and supported by Pfizer. “Pediatric Study c4591007” is an ongoing study and it was used by the FDA for approval of the vaccine for ages 5 to 12. That’s right. The FDA meeting’s data was taken from an ongoing study conducted by the very makers of the vaccine. The information below comes from the FDA Briefing Packet’s review of Pfizer Pediatric Study C4591007.
ON PAGE 39 the FDA document states (emphasis added): “Lymphadenopathy is considered an adverse reaction to vaccine and is noted as such in the EUA Fact Sheet. Among approximately 2250 children 5 to <12 years of age randomized 2:1 to receive BNT162b2 or placebo, as of the data cutoff date (06 September 2021), 13 participants (0.9%) in the BNT162b2 group and 1 participant (0.1%) in the placebo group had events of lymphadenopathy.” Lymphadenopathy is a lymph nodes disease. On page 40 the document states that “Lymphadenopathy has been identified as related to BNT162b2 in individuals ≥12 years of age and it is also observed in the pediatric 5 to <12 years of age group.” The document states that “One participant in the BNT162b2 group had a related AE of mild arthralgia (right elbow joint pain), with an onset the same day as Dose 2 (administered in the left deltoid muscle), that was reported as resolved the next day.” The document states that “One participant in the BNT162b2 group had a related AE of moderate paresthesia (bilateral lower extremity tingling) with onset at 1 day post-Dose 2 and reported as recovered/resolved 3 days after onset.” The document states that “In the BNT162b2 group, a psychiatric disorder event of tic was reported in 1 participant: One participant in the BNT162b2 group had an AE of Grade 3 tic with onset at 7 days post Dose 2 and reported as recovering/resolving at the time of the data cutoff. The AE was considered by the investigator as related to study intervention.” The document later notes that “There were 6 participants (0.4%) in the BNT162b2 group and 3 participants (0.4%) in the placebo group had events of lymphadenopathy.”
On PAGE 42 the document states that a trial participant dropped out of the trial due to an Adverse Event, stating “One participant (0.1%) in the BNT162b2 group discontinued from the vaccination period due to an AE (details in Section 3.7.2) and two participants (0.1%) in the BNT162b2 group and 1 participant (0.1%) in the placebo group withdrew from the study before the 1-month post Dose 2 visit. Neither of these withdrawals was reported as due to an AE.”
One participant had blood that passed through his/her anus, which is known as hematochezia, characterized by bloody stool. The document states that “One participant in the BNT162b2 group had a non-serious related AE reported by the investigator as moderate hematochezia 4 days after Dose 2. The participant had heme occult positive stool; was seen in the emergency department, and had no further tests done; and went home and the event resolved the same day without sequelae. This participant had a medical history of asthma and nondrug allergy and had no other reported AEs.”
On page 45, the document discusses a test subject that withdrew from the trial after developing severe pyrexia, which means severe fever, as well as severe neutropenia, which means a shortage of white blood cells, and bleeding gums. The document states that “One participant withdrew due to an AE of severe pyrexia with onset of 2 days after Dose 1 considered by the investigator as related to study intervention that resolved at 1 day after onset. Participant also had severe neutropenia (‘worsening from baseline’) with onset of 3 days after Dose 1 considered by the investigator as related to study intervention and reported as resolving at the time of the data cutoff date. Participant had a medical history of benign transient neutropenia of unknown etiology, gingivitis, and otitis media. Prior to study enrollment, she had a full hematology work-up (including for possible leukemia) with baseline absolute neutrophil count (ANC) of 480; the hematologist indicated no concerns with study participation. After Dose 1 Day 2 she reported a temperature of 40.1 °C. Her temperature returned to normal the next day. Two days after receiving Dose 1, the participant had a planned routine hematology appointment. The participant had an ANC of 20 and platelets were normal. No other symptoms or infections were reported at that time. Subsequently on Day 19 after Dose 1, the investigator was contacted by the participant’s caregiver who reported the participant had bleeding gums for 1 week prior. On Day 23, the participant attended Visit 2 to be seen by the investigator, was reported as doing well, and had a follow-up blood draw that showed the ANC had improved to 70. Dose 2 was not administered, and the participant was withdrawn from study intervention and remains in study follow-up. No other AEs were reported.”
The document states that “One participant without a reported medical history in the BNT162b2 had a related AE Type IV hypersensitivity reaction characterized by a rash on the forehead, earlobe, and right forearm 3 days after Dose 1. A dermatologist diagnosed the rash as a Type IV hypersensitivity reaction and characterized the rash as ‘plaque, erythematous and minimal crusting’, and prescribed Triamcinolone and Benadryl creams. No prohibited concomitant treatments or nonstudy vaccines had been administered. The event was reported as resolving 18 days after onset without sequelae. This participant had no other reported AEs. This participant received Dose 2 without any additional AEs reported post-dose.”
The document states that “One participant in the BNT162b2 group had a related AE of moderate angioedema reported as ‘perioral and periorbital angioedema due to allergic reaction’ and concurrent urticaria reported as ‘hives of the face and back caused due to allergic reaction’, both with onset of 2 days after Dose 2, and was reported as resolved 2 days after onset. Participant’s medical history included past allergy (hypersensitivity with mild rash) to a vaccine, Sever’s disease, contact dermatitis and seasonal allergies. This participant received no prohibited concomitant treatments or nonstudy vaccines. An analysis of angioedema cases reported in the BNT162b2 group included angioedema (see above) and urticaria (n=3). Two of the cases of urticaria were considered by the investigator as related to study intervention; one is described above (concurrent with angioedema).”
The document states that “Rashes were reported in 6 participants in the BNT162b2 group. Rashes considered as related to BNT162b2 were all mild or moderate included: rash maculo-papular, rash macular, rash papular and rash (n=1 each). Rash is considered an adverse reaction of the vaccine and is noted as such in the EUA Fact Sheet.”
On Page 47 the document details how a case of Henoch-Schoenlein purpura, a condition marked by bleeding and inflamed blood vessels, was conveniently labeled as “non related” by the vaccine makers’ clinical trial. The document states that “One participant in the BNT162b2 group had a non-serious unrelated event reported by the investigator as Henoch-Schoenlein purpura…One participant in the BNT162b2 group had a non related AE of mild Henoch Schoenlein purpura with onset of 21 days after Dose 1 and reported as ongoing at the time of the data cutoff date. The participant was treated with steroids and pain medication. Due to the initiation of steroids, the study Visit 2 appointment was delayed. This event was preceded by other non-related AEs: mild headache with onset at 10 days after Dose 1 and resolved in 2 days, and mild joint swelling of the right ankle with onset at 16 days after Dose 1 and resolved in 3 days. This participant had no reported medical history and received no prohibited concomitant treatments or nonstudy vaccines. As of the time of the data cutoff, Dose 2 was not administered.”
Negative “systemic events” and Adverse Effects were found in children between the ages of 5 and 12 and people between the ages of 16 to 25 in clinical trials of the Pfizer-BioNTech vaccine:
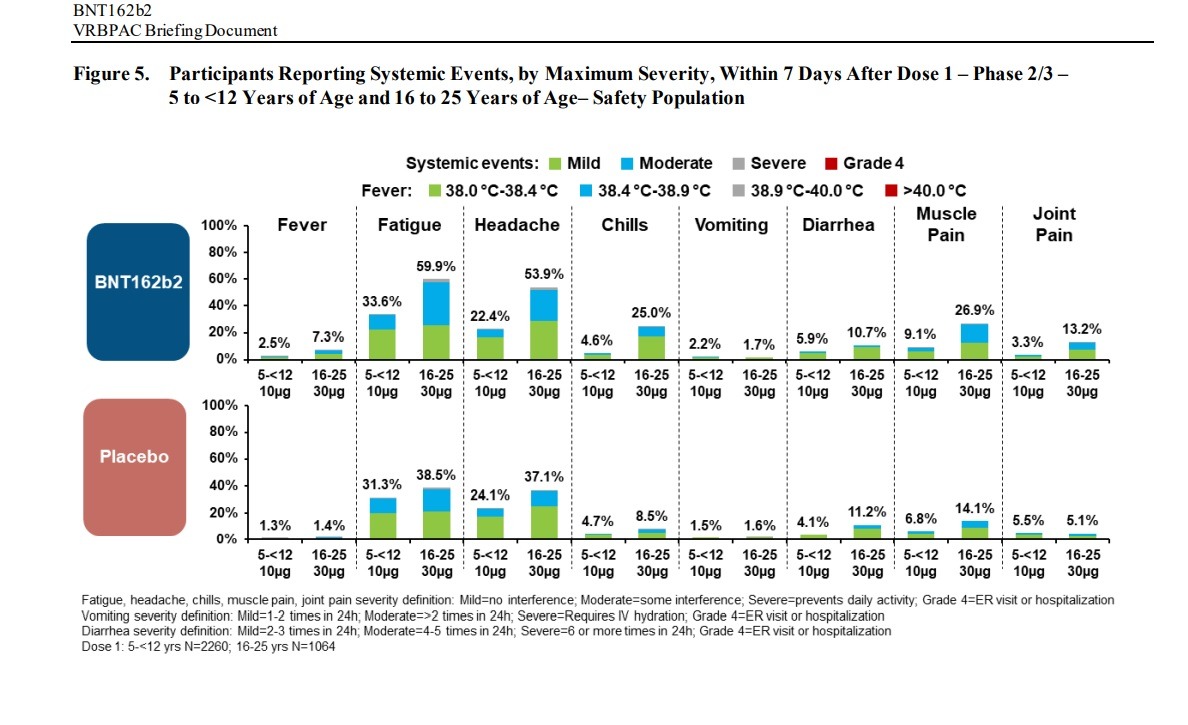
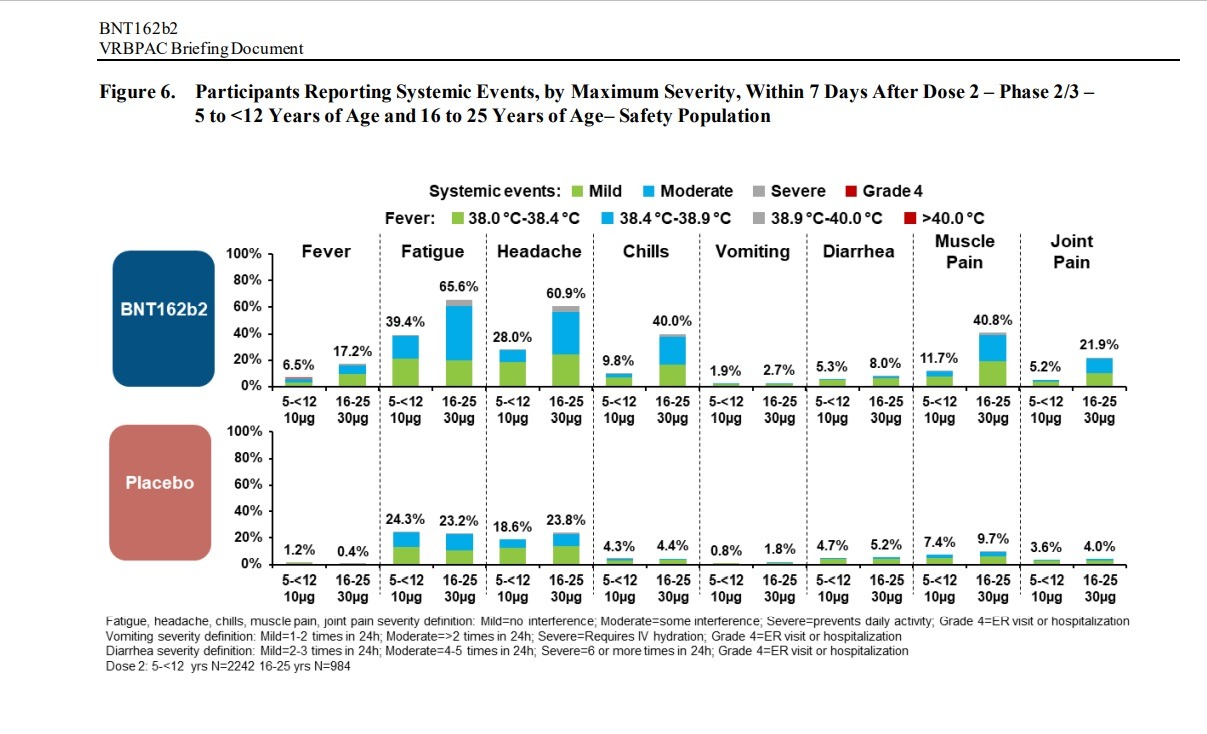
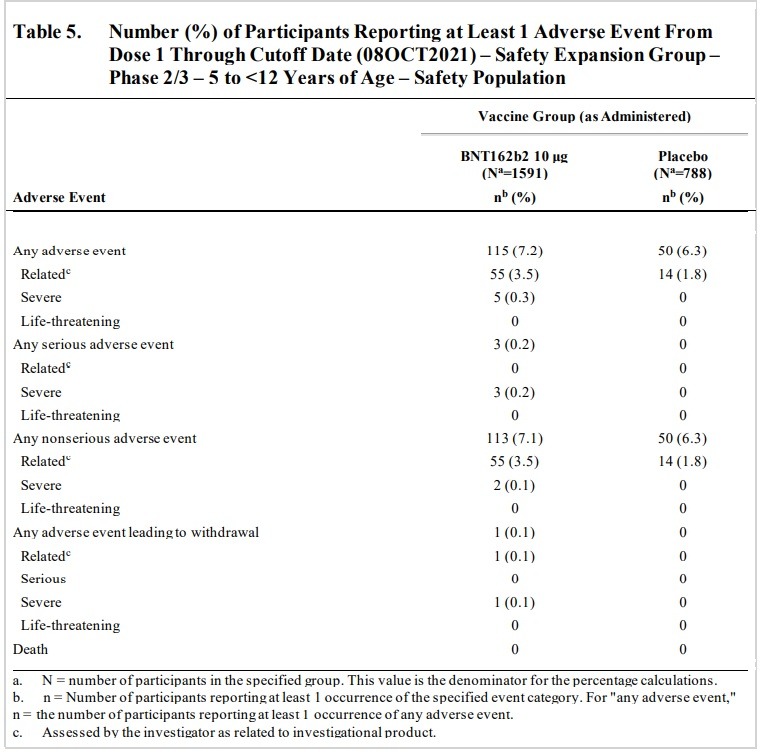
AE frequencies in these reactogenicity SOCs (BNT162b2 vs placebo)
were:
• general disorders and administration site conditions: 1.6% vs 1.7%
• gastrointestinal disorders: 1.6% vs 1.7%
• nervous system disorders: 0.7% vs 0.5%
• musculoskeletal and connective tissue disorders: 0.5% vs 0.7%
Overall, many AEs reported up to 1 month after Dose 2 were attributable to vaccine
reactogenicity events. This observation provides a reasonable explanation for the greater
rates of some AEs observed in the BNT162b2 group compared with the placebo group. In
this regard, the pattern of AEs reported in children 5 to <12 years of age was generally
consistent with that observed in prior analyses of Phase 2/3 participants ≥12 years of age in
Study C4591001.
Aside from SOCs that reflect events consistent with reactogenicity, other categories of events
are discussed below by SOC and PT. Many of the other commonly reported AEs are
consistent with events that would be expected in a general population of healthy children in
this age group and/or showed no imbalance between the vaccine and placebo groups.
• Infections and infestations were reported in 1.9% of participants in the BNT162b2 group
and 2.0% of participants in the placebo group.
• Injury, poisoning and procedural complications were reported in 1.7% of participants in
the BNT162b2 group and 0.7% of participants in the placebo group. The events reported
in this SOC are typical for this age group, such as fractures and sprains, sunburns, and
insect bites.
• Psychiatric disorders were reported in 0.3% of participants in the BNT162b2 group and
0.4% of participants in the placebo group.
• Blood and lymphatic system disorders were reported in 0.9% of participants in the
BNT162b2 group and 0.1% of participants in the placebo group, which included
lymphadenopathy and lymph node pain.
• Skin and subcutaneous disorders were reported in 1.4% of participants in the BNT162b2
group and 0.8% of participants in the placebo group, and included rashes, urticaria,
eczema, and pruritis that were overall reported more frequently in the BNT162b2 group
than in the placebo group.
Immune system disorders were reported in 0.1% of participants in the BNT162b2 group
and 0.1% of participants in the placebo group and included hypersensitivity) and other
non-drug allergies.
All of these newly reported AEs from after 2 months post-Dose 2 up to the present 3 months
post-Dose 2 were non-serious, unrelated to study intervention, and all were mild to moderate.
These newly reported AEs primarily included events consistent with mild common infections
(eg, sore throat, cough, rhinitis, enterovirus, pyrexia, fatigue) and limb fractures, which
would be expected and commonly reported in the general pediatric population.
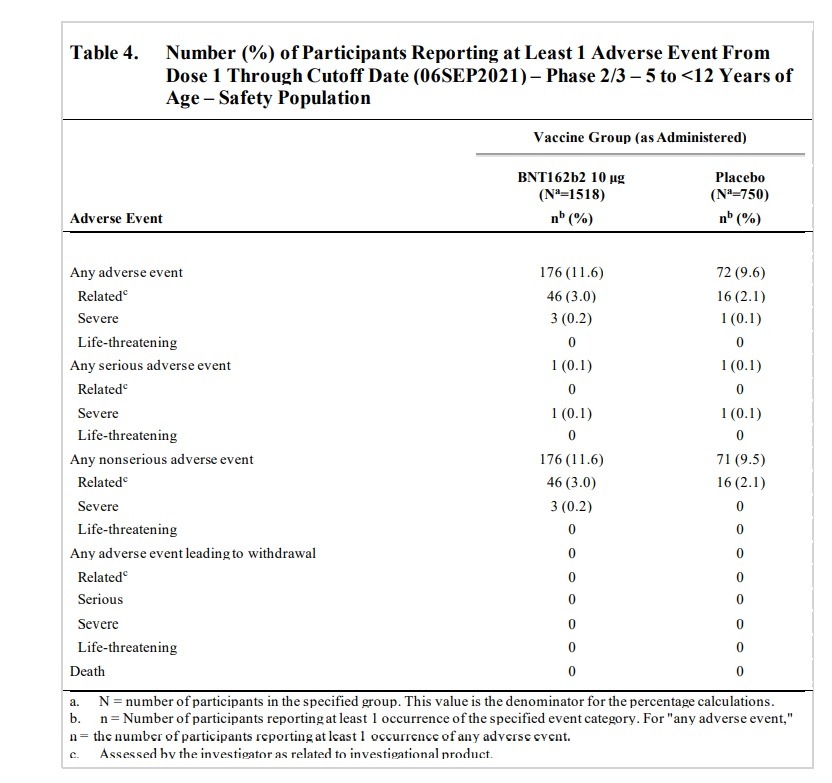
SAEs were reported from Dose 1 through the data cutoff date (06 September 2021), which
represents at least 2 months of follow-up after Dose 2. Overall, 3 SAEs were reported by
2 participants: 1 participant in the BNT162b2 group had an SAE of upper limb fracture, and
1 participant in the placebo group 2 SAEs of pancreatitis and abdominal pain (reported as
occurring ‘post-injury’) through the data cutoff date. These SAEs were all assessed by the
investigator as not related to study intervention.
Rash is considered an adverse reaction of the vaccine and is noted as such in the EUA Fact
Sheet. Overall, the pattern of events in the hypersensitivity SMQ within the skin and
subcutaneous tissue disorders SOC (including rashes) reported in children 5 to <12 years of
age in Study C4591007 was consistent with that observed in prior analyses of Phase 2/3
participants ≥12 years of age in Study C4591001.
(PAGE 39) Lymphadenopathy is considered an adverse reaction to vaccine and is noted as such in the
EUA Fact Sheet. Among approximately 2250 children 5 to <12 years of age randomized 2:1
to receive BNT162b2 or placebo, as of the data cutoff date (06 September 2021),
13 participants (0.9%) in the BNT162b2 group and 1 participant (0.1%) in the placebo group
had events of lymphadenopathy.
(PAGE 39) Additional AEs of clinical interest, regardless of inclusion on the CDC AESI list, were
evaluated based on Sponsor safety data review. These AEs were identified from the
C4591007 study database as of the data cutoff date (06 September 2021) and events
considered related by the investigator are summarized below.
Arthralgia
In the BNT162b2 group, an event of arthralgia was reported in 1 participant:
• One participant in the BNT162b2 group had a related AE of mild arthralgia (right elbow
joint pain), with an onset the same day as Dose 2 (administered in the left deltoid
muscle), that was reported as resolved the next day.
Paresthesia
In the BNT162b2 group, an event of paresthesia was reported in 1 participant:
• One participant in the BNT162b2 group had a related AE of moderate paresthesia
(bilateral lower extremity tingling) with onset at 1 day post-Dose 2 and reported as
recovered/resolved 3 days after onset.
Tic
In the BNT162b2 group, a psychiatric disorder event of tic was reported in 1 participant:
One participant in the BNT162b2 group had an AE of Grade 3 tic with onset at 7 days
post-Dose 2 and reported as recovering/resolving at the time of the data cutoff. The AE was
considered by the investigator as related to study intervention.”
(PAGE 40) Lymphadenopathy has been identified as related to BNT162b2 in individuals
≥12 years of age and it is also observed in the pediatric 5 to <12 years of age group.
Lymphadenopathy has been identified as related to BNT162b2 in study participants
≥12 years of age and is also observed in children 5 to <12 years of age, with all events
reported as mild. Rashes were more frequent in the BNT162b2 group than the placebo group,
but very few (n=4) were considered as related to vaccination and these were characterized as
mild and self-limited.
(Similarly, the overall AE and adverse reaction profile among approximately 22000
participants ≥16 years of age and 1100 adolescents 12 to 15 years of age enrolled and
vaccinated with BNT162b2 in double-blinded placebo follow-up, as of the most recent safety
cutoff date (13 March 2021), was mostly reflective of reactogenicity events with low incidences of severe and/or related events. The incidence of SAEs was low and no participants withdrew from the study due to AEs. Few deaths occurred overall in participants ≥16 years of age, and no deaths were reported in adolescents. Review of AEs of clinical interest have suggested no clear patterns or safety concerns across these studies.)
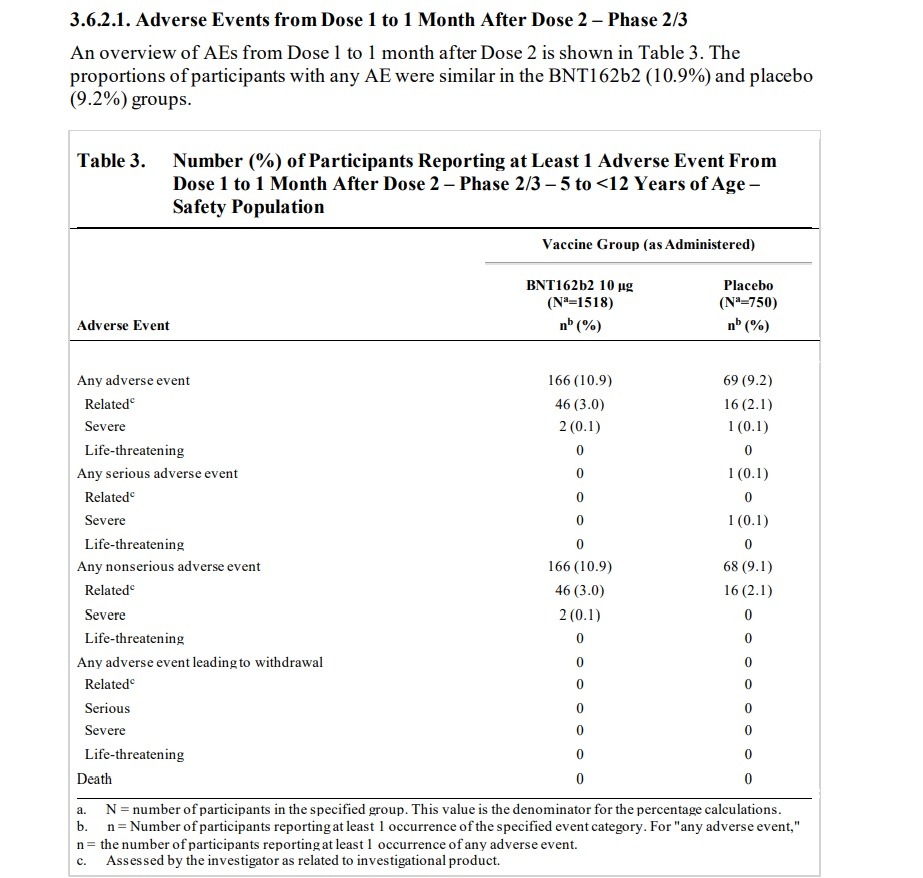
PAGE 42: One participant (0.1%) in the BNT162b2 group discontinued from the vaccination period due
to an AE (details in Section 3.7.2) and two participants (0.1%) in the BNT162b2 group and
1 participant (0.1%) in the placebo group withdrew from the study before the 1-month post Dose 2 visit. Neither of these withdrawals was reported as due to an AE
One participant in the BNT162b2 group had a non-serious related AE reported by the
investigator as moderate hematochezia 4 days after Dose 2. The participant had heme occult
positive stool; was seen in the emergency department, and had no further tests done; and went home and the event resolved the same day without sequelae. This participant had a
medical history of asthma and nondrug allergy and had no other reported AEs.
(page 45) One participant withdrew due to an AE of severe pyrexia with onset of 2 days after Dose 1
considered by the investigator as related to study intervention that resolved at 1 day after
onset. Participant also had severe neutropenia (‘worsening from baseline’) with onset of 3
days after Dose 1 considered by the investigator as related to study intervention and reported
as resolving at the time of the data cutoff date. Participant had a medical history of benign
transient neutropenia of unknown etiology, gingivitis, and otitis media. Prior to study
enrollment, she had a full hematology work-up (including for possible leukemia) with
baseline absolute neutrophil count (ANC) of 480; the hematologist indicated no concerns
with study participation. After Dose 1 Day 2 she reported a temperature of 40.1 °C. Her
temperature returned to normal the next day. Two days after receiving Dose 1, the participant
had a planned routine hematology appointment. The participant had an ANC of 20 and
platelets were normal. No other symptoms or infections were reported at that time.
Subsequently on Day 19 after Dose 1, the investigator was contacted by the participant’s
caregiver who reported the participant had bleeding gums for 1 week prior. On Day 23, the
participant attended Visit 2 to be seen by the investigator, was reported as doing well, and
had a follow-up blood draw that showed the ANC had improved to 70. Dose 2 was not
administered, and the participant was withdrawn from study intervention and remains in
study follow-up. No other AEs were reported”
“One participant without a reported medical history in the BNT162b2 had a related AE
Type IV hypersensitivity reaction characterized by a rash on the forehead, earlobe, and
right forearm 3 days after Dose 1. A dermatologist diagnosed the rash as a Type IV
hypersensitivity reaction and characterized the rash as ‘plaque, erythematous and
minimal crusting’, and prescribed Triamcinolone and Benadryl creams. No prohibited
concomitant treatments or nonstudy vaccines had been administered. The event was
reported as resolving 18 days after onset without sequelae. This participant had no other
reported AEs. This participant received Dose 2 without any additional AEs reported
post-dose.
• One participant in the BNT162b2 group had a related AE of moderate angioedema
reported as ‘perioral and periorbital angioedema due to allergic reaction’ and concurrent
urticaria reported as ‘hives of the face and back caused due to allergic reaction’, both
with onset of 2 days after Dose 2, and was reported as resolved 2 days after onset.
Participant’s medical history included past allergy (hypersensitivity with mild rash) to a
vaccine, Sever’s disease, contact dermatitis and seasonal allergies. This participant
received no prohibited concomitant treatments or nonstudy vaccines.
An analysis of angioedema cases reported in the BNT162b2 group included angioedema (see
above) and urticaria (n=3). Two of the cases of urticaria were considered by the investigator
as related to study intervention; one is described above (concurrent with angioedema)”
“Rashes were reported in 6 participants in the BNT162b2 group. Rashes considered as related
to BNT162b2 were all mild or moderate included: rash maculo-papular, rash macular, rash
papular and rash (n=1 each). Rash is considered an adverse reaction of the vaccine and is
noted as such in the EUA Fact Sheet.”
(PAGE 47) One participant in the BNT162b2 group had a non-serious unrelated event reported by the
investigator as Henoch-Schoenlein purpura.
• One participant in the BNT162b2 group had a non-related AE of mild HenochSchoenlein purpura with onset of 21 days after Dose 1 and reported as ongoing at the
time of the data cutoff date. The participant was treated with steroids and pain
medication. Due to the initiation of steroids, the study Visit 2 appointment was delayed.
This event was preceded by other non-related AEs: mild headache with onset at 10 days
after Dose 1 and resolved in 2 days, and mild joint swelling of the right ankle with onset
at 16 days after Dose 1 and resolved in 3 days. This participant had no reported medical
history and received no prohibited concomitant treatments or nonstudy vaccines. As of
the time of the data cutoff, Dose 2 was not administered.
Lymphadenopathy
There were 6 participants (0.4%) in the BNT162b2 group and 3 participants (0.4%) in the
placebo group had events of lymphadenopathy.
It's not often you see something that really resonate with you as much as this did.
You see, 7 years ago I was diagnosed with Henoch Schönlein's Purpura:
This is an illness that's most often seen in the age of 4-11. I got it when i was 16 years old. 1/7 https://t.co/Uyy0sBzG0D— Noel Rose (𝘩𝘦𝘯𝘱𝘢𝘪) (@PlasticTasticc) October 28, 2021
The Pfizer vaccine trial:
C4591007
(page 47)

There were 6 participants (0.4%) in the BNT162b2 group and 3 participants (0.4%) in the placebo group had events of lymphadenopathy
(page 39) Arthralgia In the BNT162b2 group, an event of arthralgia was reported in 1 participant: One participant in the BNT162b2 group had a related AE of mild arthralgia (right elbow joint pain), with an onset the same day as Dose 2 (administered in the left deltoid muscle), that was reported as resolved the next day.
Paresthesia In the BNT162b2 group, an event of paresthesia was reported in 1 participant: One participant in the BNT162b2 group had a related AE of moderate paresthesia (bilateral lower extremity tingling) with onset at 1 day post-Dose 2 and reported as recovered/resolved 3 days after onset.
Tic In the BNT162b2 group, a psychiatric disorder event of tic was reported in 1 participant: One participant in the BNT162b2 group had an AE of Grade 3 tic with onset at 7 days post-Dose 2 and reported as recovering/resolving at the time of the data cutoff. The AE was considered by the investigator as related to study intervention.
mild to moderate redness and swelling occurred at higher frequencies in children than previously reported in C4591001. Systemic events most commonly included fatigue, headache, and muscle pain, and generally increased in frequency and/or severity with increasing dose number; these were typically milder and less frequent than previously reported in Study C4591001
Lymphadenopathy has been identified as related to BNT162b2 in study participants ≥12 years of age and is also observed in children 5 to <12 years of age, with all events reported as mild. Rashes were more frequent in the BNT162b2 group than the placebo group, but very few (n=4) were considered as related to vaccination and these were characterized as mild and self-limited
One participant (0.1%) in the BNT162b2 group discontinued from the vaccination period due to an AE (details in Section 3.7.2) and two participants (0.1%) in the BNT162b2 group and 1 participant (0.1%) in the placebo group withdrew from the study before the 1-month postDose 2 visit. Neither of these withdrawals was reported as due to an AE.
One participant withdrew due to an AE of severe pyrexia with onset of 2 days after Dose 1
considered by the investigator as related to study intervention that resolved at 1 day after
onset. Participant also had severe neutropenia (‘worsening from baseline’) with onset of 3
days after Dose 1 considered by the investigator as related to study intervention and reported
as resolving at the time of the data cutoff date. Participant had a medical history of benign
transient neutropenia of unknown etiology, gingivitis, and otitis media. Prior to study
enrollment, she had a full hematology work-up (including for possible leukemia) with
baseline absolute neutrophil count (ANC) of 480; the hematologist indicated no concerns
with study participation. After Dose 1 Day 2 she reported a temperature of 40.1 °C. Her
temperature returned to normal the next day. Two days after receiving Dose 1, the participant
had a planned routine hematology appointment. The participant had an ANC of 20 and
platelets were normal. No other symptoms or infections were reported at that time.
Subsequently on Day 19 after Dose 1, the investigator was contacted by the participant’s
caregiver who reported the participant had bleeding gums for 1 week prior. On Day 23, the
participant attended Visit 2 to be seen by the investigator, was reported as doing well, and
had a follow-up blood draw that showed the ANC had improved to 70. Dose 2 was not
administered, and the participant was withdrawn from study intervention and remains in
study follow-up. No other AEs were reported
Psychiatric disorders were reported in 1 participant in the BNT162b2 group (a related
event of ‘irritability’) and none in the placebo group.
• Blood and lymphatic system disorders were reported in 0.4% of participants in the
BNT162b2 group and 0.4% of participants in the placebo group, which included
lymphadenopathy.
• Skin and subcutaneous disorders were reported in 1.0% of participants in the BNT162b2
group and 0.5% of participants in the placebo group. Events reported more frequently in
the BNT162b2 group included rashes, urticaria, angioedema, dermatitis, pruritis, night
sweats.
• Immune system disorders were reported in 3 participants (0.2%) in the BNT162b2 group
and none in the placebo group and included one event reported as Type IV
hypersensitivity reaction and other non-drug allergies.
One participant in the BNT162b2 group had a non-serious related AE reported by the investigator as moderate hematochezia 4 days after Dose 2. The participant had heme occult positive stool; was seen in the emergency department, and had no further tests done; and went home and the event resolved the same day without sequelae. This participant had a medical history of asthma and nondrug allergy and had no other reported AEs
One participant without a reported medical history in the BNT162b2 had a related AE
Type IV hypersensitivity reaction characterized by a rash on the forehead, earlobe, and
right forearm 3 days after Dose 1.
One participant in the BNT162b2 group had a related AE of moderate angioedema
reported as ‘perioral and periorbital angioedema due to allergic reaction’ and concurrent
urticaria reported as ‘hives of the face and back caused due to allergic reaction’, both
with onset of 2 days after Dose 2, and was reported as resolved 2 days after onset.
Participant’s medical history included past allergy (hypersensitivity with mild rash) to a
vaccine, Sever’s disease, contact dermatitis and seasonal allergies. This participant
received no prohibited concomitant treatments or nonstudy vaccines
via IFTTT
InoreaderURL: SECONDARY LINK